

Fundamental concepts of vibrational spectroscopy For IIT JAM is a crucial topic in physical chemistry that deals with the interaction of matter with electromagnetic radiation, and its applications in identifying molecular structures and properties.
Syllabus – Physical Chemistry for IIT JAM
In standard prep setups, this topic sits comfortably inside Unit 2: Physical Chemistry of the IIT JAM syllabus.
If you want to look it up in standard textbooks, you can flip open Physical Chemistry by P W Atkins (Chapter 6 is your goldmine here). Another fantastic reference that clears up the math without making your head spin is Physical Chemistry: A Molecular Approach by Donald A. McQuarrie and John D. Simon. Here at VedPrep, we always tell our students that spending a little time with these reference books can completely change how you look at the subject.
Key areas to lock down for the exam include the core principles of infrared (IR) and Raman spectroscopy, figuring out molecular vibrations, and learning how to actually read a spectrum without guessing.
Fundamental concepts of vibrational spectroscopy For IIT JAM – Introduction
Let’s break this down into plain English. Vibrational spectroscopy is just the study of how molecules absorb or scatter energy in the infrared (IR) and Raman regions of the light spectrum. Think of it as a fingerprint scanner for molecules. By looking at how a molecule vibrates, you can tell exactly what bonds it has and how they are arranged.
You will mostly deal with two main types: Infrared (IR) spectroscopy and Raman spectroscopy.
-
IR Spectroscopy: This happens when a molecule directly swallows a photon of infrared light.
-
Raman Spectroscopy: This is more about scattering. Light hits the molecule, bounces off, and loses or gains a bit of energy along the way.
They are like two different camera angles of the same building—you need both to see the whole picture.
By using vibrational spectroscopy, you get a clean snapshot of its molecular structure. Whether you are dealing with a simple diatomic molecule or a messy organic chain, this technique is your go-to tool. For an IIT JAM aspirant, mastering the different types of vibrational modes, the selection rules, and spectral interpretation is what stands between an average rank and a top-IIT seat.
Fundamental concepts of vibrational spectroscopy For IIT JAM
Now, let’s look at the actual physics behind the magic. You might have studied the ideal harmonic oscillator in physics—where a bond acts like a perfect spring. But real life isn’t perfect. Molecules follow the anharmonic oscillator model.
Imagine a rubber band. If you stretch it a little, it snaps back. But if you pull it too hard, it loses its bounce or snaps entirely. That is what anharmonicity is. The potential energy curve warps and flattens out as the atoms get pulled too far apart, which changes the energy gaps between levels.
Molecules can move in a couple of basic ways: stretching (bonds getting longer and shorter) and bending (angles changing). As they vibrate, they usually rotate too. This rotational twisting messes with the energy levels, giving you those fine lines you see on a real spectrum.
The big takeaway here is the quantization of vibrational energy. Just like stepping up a ladder, a molecule cannot hold just any random amount of vibrational energy; it has to jump between specific, discrete steps. We use the Schrödinger equation to calculate these exact energy steps, factoring in the imperfections of our “imperfect spring” (anharmonicity). Getting a firm grip on these quantum rules is exactly how you crack the tricky numerical questions in the IIT JAM exam.
Worked Example – Vibrational Spectroscopy
Let’s look at a classic problem you might see on test day.
Problem: A unknown liquid with the formula C3H6O goes through an IR scanner. The spectrum shows a massive, sharp peak at 1720 cm-1 and a weaker, smaller peak near 2800 cm-1. What is the structure, and does the math check out for the main bond?
Step 1: Analyze the Peaks
That huge peak at 1720 cm-1 is a dead giveaway for a carbonyl group (C=O). The weaker signal around 2800 cm-1 is typically due to a C-H stretch. Because there is no broad O-H blob or an aldehyde-specific double peak, we are likely looking at a ketone. With three carbons, our prime suspect is propanone (acetone), CH3COCH3.
Step 2: Verification via Hooke’s Law
Let’s treat the C=O bond like a spring to see if the math matches the machine. We use the wavenumber formula derived from Hooke’s Law:
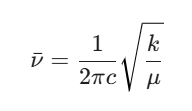
Where:
-
k is the bond force constant (for a double bond like C=O, it is quite stiff, around 1200 N/m to 1850 N/m. Let’s use 1850 N/m.
-
c is the speed of light (3 × 1010 cm/s).
-
μ is the reduced mass of the C and O atom pair.
Let’s quickly calculate the reduced mass μ:.
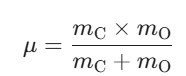
Using standard atomic masses (12 amu and 16 amu) and converting to kilograms gives us roughly 1.14 × 10-26 kg.
Plugging k = 1850 N/m and μ = 1.14 × 10-26 kg into our formula, the calculated frequency comes out right around 1720 cm-1. The math matches the experimental data perfectly, confirming our molecule is indeed acetone.
Common Misconceptions in Fundamental concepts of vibrational spectroscopy For IIT JAM
As per vibrational spectroscopy, a very common trap IIT JAM aspirants fall into is thinking IR and Raman spectroscopy are basically the same thing since they both look at vibrations. That is a quick way to lose negative marks.
The secret lies in the selection rules:
-
For a vibration to show up in an IR spectrum, the molecule’s dipole moment must change during the vibration.
-
For it to show up in a RAMAN spectrum, the molecule’s polarizability must change.
Think of it like this: Imagine a fictional scenario where two friends are testing a new sports car. One friend only cares about how fast the car can accelerate (that’s IR checking for dipole changes). The other friend only cares about how well the car handles tight corners (that’s Raman checking for shape deformability, or polarizability). They are looking at the exact same car, but measuring completely different traits.
Because of this, highly polar bonds (like O-H or C=O) scream out loudly in IR, while symmetric, non-polar bonds (like C=C or N≡N) prefer to show off in Raman.
Another mistake is ignoring the solvent. Students often think the solvent is just a passive liquid. In reality, if your solvent forms hydrogen bonds with your sample, it will drag your peak positions around and change how sharp they look.
Finally, remember that vibrational spectroscopy isn’t a silver bullet. It tells you what functional groups you have, but it struggles to tell you exactly how the whole carbon skeleton is glued together.
| Technique | Main Limitation |
| Vibrational Spectroscopy | Overlapping peaks, messy mixtures, and ambiguity in exact structural mapping |
Applications of Fundamental concepts of vibrational spectroscopy For IIT JAM
Why should you care about vibrational spectroscopy beyond clearing the exam? Because it is used everywhere in modern science.
In chemical labs, Fourier Transform Infrared (FTIR) spectroscopy is the absolute gold standard for checking if a reaction worked or if a polymer sample is pure. If a pharmaceutical company is making a batch of aspirin, they run it through an IR scan to ensure no unreacted starting materials are left behind.
Material scientists use Raman spectroscopy to study cutting-edge materials like graphene and carbon nanotubes. Because Raman is incredibly sensitive to symmetrical crystal structures, it can tell researchers exactly how many atomic layers of graphene are stacked in a sample just by how the light scatters.
-
Medicine: Identifying anomalies in tissue or verifying drug purity.
-
Forensics: Checking unknown powders or paint chips at a scene.
-
Environment: Tracking trace pollutants in water and air.
Understanding the core setup—from vibrational spectroscopy to how light splits inside the machine—is what helps you make sense of these applications when they pop up in application-based exam questions.
Exam Strategy for Fundamental concepts of vibrational spectroscopy For IIT JAM
When the exam date starts getting closer, your study strategy needs to get sharp. You cannot just memorize formulas; you need to know when to apply them.
As per vibrational spectroscopy, focus heavily on predicting whether a molecular vibration is IR-active, Raman-active, or both. This is where a little bit of molecular symmetry and group theory pays off big time. Keep a cheatsheet of characteristic IR stretching frequencies (O-H, N-H, C=O, C-C) so you can rapidly identify molecules during the exam.
We at VedPrep recommend breaking your preparation down into a clear checklist:
-
Nailed down the basics: Do you truly understand the difference between a harmonic and an anharmonic oscillator?
-
Problem-solving loops: Are you practicing numerical questions on reduced mass and force constants?
-
Mock analysis: Are you taking targeted subject tests to check your speed?
Using structured study materials, like the deeply researched question banks we print at VedPrep, can save you hours of wandering through irrelevant topics.
Conclusion
Vibrational spectroscopy might seem intimidating with its mix of quantum mechanics, spring equations, and selection rules, but it is one of the most logical and rewarding topics in the IIT JAM physical chemistry syllabus. Once you look past the dense textbook language and see it as a simple tool that tracks molecular movement, the entire subject opens up. Focus on mastering the difference between IR and Raman, practice your Hooke’s law calculations, and learn to read spectral peaks like clues in a puzzle.
To learn more in detail from our faculty, watch our YouTube video:
Frequently Asked Questions
Where does vibrational spectroscopy fit within the IIT JAM syllabus?
It is a major component of Physical Chemistry under the Molecular Spectroscopy unit. It frequently connects with other core chemistry topics like chemical bonding, quantum mechanics, and organic structure determination.
What are the two main types of molecular vibrations?
Molecules primarily undergo stretching vibrations (where the bond length continuously changes along the bond axis) and bending vibrations (where the angle between two bonds changes).
Why does a typical vibrational spectrum look like a series of bands rather than sharp single lines?
Because vibrational transitions do not happen in isolation. When a molecule jumps to a higher vibrational energy level, it simultaneously undergoes transitions between closer spaced rotational energy levels, which blurs the sharp lines into broader bands.
Which standard reference textbooks are best for studying this topic for IIT JAM?
You can safely rely on Physical Chemistry by P.W. Atkins for a strong conceptual base, and Physical Chemistry: A Molecular Approach by McQuarrie & Simon to master the quantum mechanical derivations and numerical problems.
Is a molecule with zero dipole moment completely inactive in IR spectroscopy?
Not necessarily. While the molecule as a whole might be non-polar at rest (CO2), individual asymmetric vibrations can induce a temporary dipole moment. If a specific vibration creates a change in the dipole moment, that specific mode will be IR active.
Can we assume that a stiffer bond always absorbs at a higher wavenumber?
Generally yes, because wavenumber is directly proportional to the square root of the force constant (√k). However, you must always look at the reduced mass (μ) as well. If the atoms are incredibly heavy, they can counteract a high force constant.
What is a common pitfall when differentiating between IR and Raman activity?
Students often misapply the rule of mutual exclusion. This rule only applies to molecules that possess a center of inversion (like CO2 or trans-dichloroethylene). For these molecules, IR active modes are Raman inactive, and vice versa. If there is no center of inversion, a mode can easily be active in both.
Why shouldn't you rely solely on vibrational spectroscopy to deduce an unknown organic structure?
Vibrational spectroscopy is excellent at spotting functional groups (like a C=O or an O-H stretch), but it cannot tell you how the carbon skeleton is branched or where a functional group is positioned along a chain. For complete identification, you must combine it with NMR and mass spectrometry.
How do solvent interactions alter spectral results?
If you use a polar solvent that forms hydrogen bonds with your sample, it will weaken the target bond, causing its force constant (k) to drop. This shifts your expected absorption peak to a lower wavenumber and can broaden the peak significantly.
What is the fundamental difference between a harmonic and an anharmonic oscillator model?
In a harmonic oscillator, the potential energy curve is a perfect parabola, meaning a bond can be stretched infinitely without breaking, and energy levels are equally spaced (ΔE is constant). The anharmonic model (like the Morse potential) accounts for real-life bond dissociation; the curve flattens out at large distances, and the energy gaps get progressively narrower as you go higher.
What are Rayleigh, Stokes, and Anti-Stokes scattering in Raman spectroscopy?
-
Rayleigh Scattering: The light bounces off without changing energy (elastic scattering).
-
Stokes Scattering: The scattered light loses energy to the molecule, coming out at a lower frequency.
-
Anti-Stokes Scattering: The light hits an already excited molecule, absorbing its energy and coming out at a higher frequency.
Why are Stokes lines typically much more intense than Anti-Stokes lines?
According to the Maxwell-Boltzmann distribution, at standard room temperatures, the vast majority of molecules rest in their lowest vibrational ground state (v=0). Since Anti-Stokes scattering requires the molecule to already be in an excited state (v=1), it happens far less frequently, resulting in much weaker peaks.







